HomozygosityMapper
Technical documentation
| database schema | web interface |
Click image to enlarge.
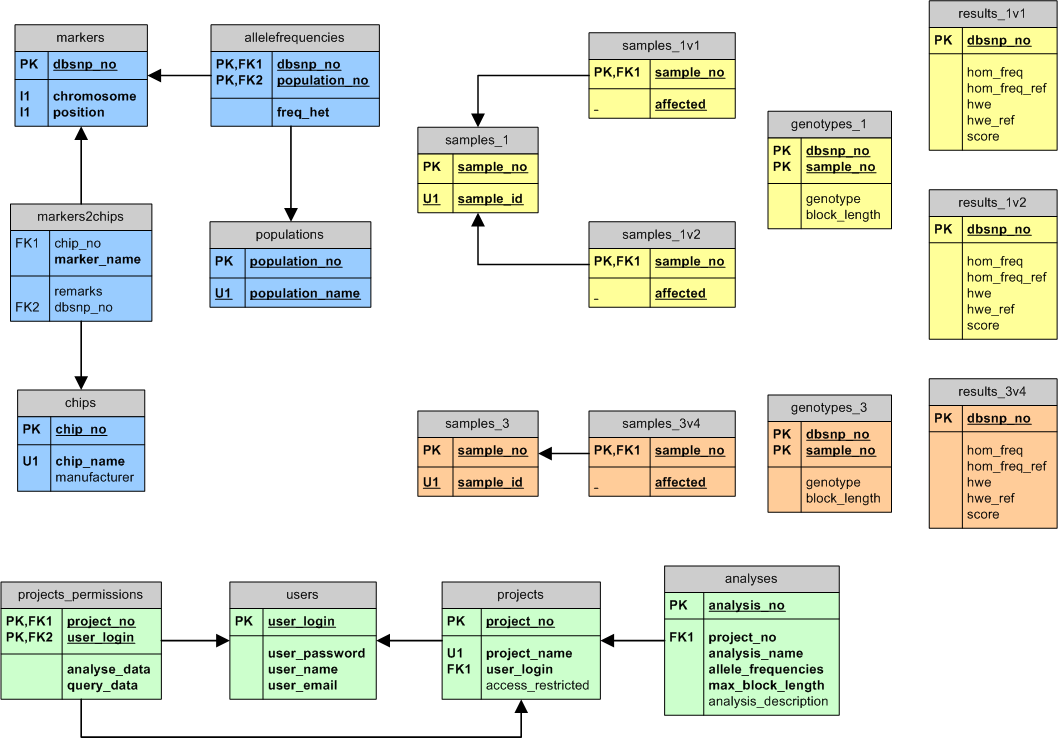
HomozygosityMapper
uses a very simple, strictly relational database schema. Besides a
group of administrative tables (green), the database is split into two
distinct parts: marker data (light blue) and project data. Each project
is stored in its own tables reducing table sizes and hence access
times; this also facilitates the deletion of single projects. To speed
up data input, the relations within a project are not connected by
foreign key constraints - since all data processing is done by the
application, referential integrity can be guaranteed without explicit
foreign keys.
The 'markers' table is shared with GeneDistiller and MutationTaster.
| data analysis | web interface |
In a first step, conducted directly after data upload, HomozygosityMapper detects homozygous stretches in all samples. Pseudo-code example:
for sample (samples){
for chromosome (chromosome){
get genotypes from DB ordered by position (chromosome, sample)
as array (index: pos, value: genotype)
while (pos <= number of elements (genotypes)) {
if (genotypes[pos]=='heterozygous'){
blocklength[pos]=0;
pos++;
}
else {
pos2=pos;
while (! DetectBlockEnd(pos,pos2) && pos2 <= number of elements (genotypes)){
pos2++;
}
blocklength=pos2-pos;
for my pos3 (pos..(pos2-1)){
blocklength[pos3]=pos3;
}
pos=pos;
}
}
}
}
The function DetectBlockEnd (which is not shown here) returns true when a genotype is heterozygous and not neighboured by 7 or more homozygous/unknown genotypes on each side. Although this significantly slows down block detection, it renders HomozygosityMapper more robust against genotyping errors; which may occur with a frequency of 2% with DNA of low quality or inappropriate genotype calling settings.
The choice of 7 markers reflects spacing and error rate (which increases on Affymetrix high-density arrays).
Homozygosity scores in the conventional model
The second step is the user-defined data analysis in which cases and controls can be specified. Also, the users can enter a block length limit - higher block lengths will be set to this value when calculating the homozygosity score. This prevents inflation of homozygosity scores by very long homozygous stretches in single individuals. As default, values optimal for the marker density are used to limit block lengths.
Score calculation is quite simple:
HomScore (marker) = SUM (samples) (BlockLength (marker, sample))
In pseudo-code:
for marker (markers){
score[marker]=0;
for sample (samples){
if (blocklength[sample] > maximum_block_length){
score[marker]+=maximum_block_length;
}
else {
score[marker]+=blocklength[sample];
}
}
}
Homozygosity scores in the 'genetic homogeneity' model
This model uses a far more complex approach than scoring homozygous blocks only and hence the analysis takes considerably longer.
In a first step, the 'reference homozygous' genotype is calculated for each marker. This is always the most frequent homozygous genotype in the cases. In the second step, the control samples are checked for the presence of this genotype. If, in any of the samples, a long stretch of the respective 'reference homozygosity' is found, this region is excluded. This block length can be adjusted in the analysis settings - we recommend to use high values to reduce the risk of false negatives due to long stretches of uninformative SNPs.
In the last step, the case samples are studied. When they share the 'reference homozygosity', the length of the shared segment is added to the homozygosity score (as long as the region is not excluded due to the controls). Here, 'false' genotypes are tolerated to a certain amount. Whenever a single genotype does not match the 'reference homozygous' genotype (i.e. homozygous for the other allele or heterozygous) but is surrounded by 6 correct geneotyps on both sides, it is neglected. As in the normal model, unknown genotypes are counted as reference homozygous in the cases. In the controls, they are not treated as reference homozygous, again to reduce the risk of false negatives.
Please note that the recommended values are quite high. Depending on your families' structures, you might wish to lower it significantly. You should, however, keep in mind that this will increase the risk of false negatives, so please be careful.